Proteine für die Krebstherapie dank Programmierkniff
Schnellere Energieberechnungen beim Proteindesign
Neue Proteine für biomedizinische oder andere Anwendungen computergestützt zu entwickeln, erfordert lange Rechenzeiten auf leistungsstarken Servern. Ein Team aus Forschenden des Max-Planck-Instituts für Biologie Tübingen und des Universitätsklinikums Tübingen hat nun eine neue Berechnungsmethode entwickelt und getestet, die die notwendigen Energiekalkulationen erheblich beschleunigt. Ihr Ansatz ermöglicht eine präzise und effiziente Entwicklung von Funktionsproteinen. Die Forschenden demonstrierten darüber hinaus den Nutzen ihrer Ergebnisse, indem sie zwei Klassen von Proteinen entwickelten, die in der Krebsdiagnostik und -therapie eingesetzt werden können.

Proteine sind in vielerlei Hinsicht lebenswichtig: Manche sorgen für die Festigkeit von Haaren und Knochen, andere regulieren den Stoffwechsel oder erkennen und bekämpfen Viren – um nur ein paar ihrer vielfältigen Aufgaben zu nennen. Daher hat die Entwicklung neuer Proteine mit speziellen Funktionen das Potential, die Biomedizin zu revolutionieren.
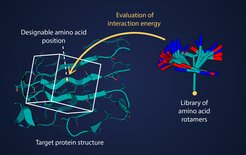
Wenn Forschende ein Protein mit einer gewünschten Funktion entwerfen, legen sie zunächst dessen räumliche Gesamtstruktur fest, denn die Form bestimmt, wie das Protein mit anderen Molekülen interagiert. Die Herausforderung besteht dann darin, eine stabile Anordnung der Bausteine zu finden, aus denen sich die Zielstruktur zusammensetzen kann. Diese Bausteine sind Aminosäuren: organische Moleküle, die zu langen Ketten aneinandergereiht die Proteine ergeben. Die Interaktion der Aminosäuren und all ihrer Atome legt die dreidimensionale Gestalt eines Proteins und somit seine Funktion fest.
Drastische Reduzierung des Rechenaufwands
Da die Abfolge der Aminosäuren im Protein die Form bestimmt, können schon kleine Änderungen – etwa der Austausch einer einzelnen Aminosäure gegen eine andere – die erwünschte Struktur beträchtlich stabilisieren oder destabilisieren. „Um ein stabiles Protein zu entwickeln, muss man die Energie unzähliger Aminosäuresequenzen berechnen und vergleichen“, erklärt Mohammad ElGamacy, Forschungsgruppenleiter am Universitätsklinikum Tübingen. „Unter rechnerischen Gesichtspunkten sind diese Kalkulationen sind das Nadelöhr fürs Proteindesign.“
Daher haben ElGamacy und seine Kollaborationspartnerinnen und -partner nun ein neues Berechnungsverfahren entwickelt, das nicht nur die Energiekalkulation um Größenordnungen beschleunigt, sondern auch grundlegende Verbesserungen der Genauigkeit ermöglicht. Sie haben als Erste das Verfahren der sogenannten Tensorierung auf das Problem des Proteindesigns angewendet: eine Methode, die für Grafik-Rendering und Computerspiele schon intensiv genutzt wird.
Statt die Wechselwirkungsenergie für jedes Paar räumlich naher Atome einzeln zu berechnen, ermitteln sie vorab den Großteil der Interaktionsinformation für jede der Aminosäuren und speichern diese in einem dreidimensionalen Raster. Mit diesen Rastern kann man dann die Wechselwirkungsenergien für jeden Einzelfall unaufwändig berechnen. „Uns freut besonders, dass die Beschleunigung der Energieberechnungen ohne Maschinelles Lernen möglich war“, sagt Kateryna Maksymenko, die in der Abteilung für Proteinevolution des Max-Planck-Instituts für Biologie Tübingen forscht und Hauptautorin der Studie ist. „Unsere Kalkulationen basieren rein auf Physik. Daher können wir immer nachvollziehen, wie und warum die Software ihre Ergebnisse erzielt hat. Mit einem Deep-Learning-Algorithmus wäre das unmöglich.”
Klinische Anwendungen in Krebsdiagnostik und -therapie
Den Nutzen ihrer Methode demonstrierten die Forschenden gleich mit: Sie entwickelten zwei Klassen von Proteinen und testeten diese in Zusammenarbeit mit Forschenden des Universitätsklinikums Tübingen und des Werner Siemens Imaging Center. Die erste der beiden Erfindungen blockiert die Kommunikation des EGF-Rezeptors, eines Proteins, das Zellwachstum und -differenzierung regelt und eine wichtige Rolle bei verschiedenen Krebsarten spielt.
Das zweite neuentwickelte Protein ist ebenfalls für den direkten klinischen Einsatz geeignet. „Besonders daran ist die mögliche Zweifachwirkung in Krebsdiagnostik und -therapie“, hebt Maksymenko hervor. „Das Protein bindet an Kupfer-64; das heißt, seine Verteilung im Körper kann nuklearmedizinisch nachverfolgt werden. Doch es kann ebenfalls das Radioisotop Kupfer-67 transportieren, das die Krebszellen tötet.“ Hinzu kommt, dass das Protein an 13 Kupferionen gleichzeitig binden kann; diese bislang unerreichte Dichte macht das Molekül besonders geeignet für Bildgebung in höchster Qualität.
In einem weiteren Entwicklungsschritt möchte das Team den Kupferliganden mit anderen therapeutisch relevanten Proteinen verbinden und deren Ausbreitung im Körper untersuchen. Die Forschenden sind überzeugt, dass ihr Ansatz bei der Entwicklung vieler weiterer Proteine unterstützen wird.